What changes when a drug candidate moves from the research stage to GMP production – and why?
The whys and the whats of GMP production of biopharmaceuticals.
Why’d you have to go and make things so complicated? This chorus of the pop-rock song, Complicated, may reflect the thoughts of drug developers when their first project moves to clinical production. Biopharmaceutical production must follow Good Manufacturing Practice, GMP, which is a system for ensuring that products are constantly produced and controlled according to quality standards. The difference between the research stage and GMP production is striking – even confusing. Why does GMP exist? What does it mean in practice?
To answer these questions, we turned to Bio-CDMO experts Magnus Gustafsson and Eero Mustalahti. Here they review the history of events that led to the development of GMP and discuss how the operational environment of a GMP manufacturing site differs from that of a research laboratory.

History of events towards the development of GMP
“Pharmaceutical regulations are there for a good reason,” Gustafsson says. “In the early 1900s, traveling charlatans were selling bottles of miracle elixir that was said to be good for anything from aches and rheumatism to the cure of cancer. Today it feels unreal, but only a 100 years ago alcohol, opium, or morphine could be included in medicines to calm babies with colic ” As late as 1912, the International Hague Convention forced governments to implement legislation that effectively reduced access to opium and broke the dangerous habit.”
“Manufacturing technology was also primitive. In the early 1900s, serum extracted from an old milk wagon horse named Jim was used to produce antibodies against diphtheria toxin. Jim had been dosed with diphtheria toxin and its serum, containing those antibodies was used as a treatment of the disease. Unfortunately, Jim also got infected with tetanus. Thirteen children died of the contaminated injection. Around the same time, several children died after receiving a contaminated smallpox vaccine. There was a real Wild West in medicine and no regulations to protect the public from dangerous products.”
“The Federal Food, Drug, and Cosmetic Act, a precursor to GMP and FDA, was passed in 1938 in response to a cough medicine tragedy that caused mass poisoning, killing over 100 people. A chemist had used diethylene glycol, used in antifreeze, to dissolve the sulfanilamide into a syrup making the drug easier to take. As no law required safety studies of new drugs, the chemist had failed to note that the new formulation was a deadly poison. The Federal Food, Drug, and Cosmetic Act requested that drug manufacturers show that the drug is safe before marketing it. But history repeated itself in 2007 when diethylene glycol containing cough syrup caused the death of 365 people in Panama. The imported diethylene glycol from a Chinese manufacturer was sold under the name TD glycerine, which means ‘glycerine substitute’. A Spanish middleman filling the customs declaration changed the name to glycerine. Using today’s GMP guidelines in the EU and US this mix-up should not be possible since GMP specifies identity control of all used raw materials.”
“Unfortunately, the 1938 act in the US did not prevent the Thalidomide tragedy from happening in Europe in the late 1950s. The medicine produced in Germany was used as a sleeping pill and it became a popular off-label medicine against morning sickness during pregnancy. The drug was banned in most countries by the early 1960s after several children were born with severe birth defects. Subsequently, the Kefauver Harris Drug Amendment was signed by President Kennedy in 1962. The amendment, for the first time, required drug manufacturers to prove that the medicine is not only safe but also effective. The regulation of clinical studies was created, as well as procedures for regulatory approvals and quality assurance in the development of drug products.”
The first GMP
“Public pressure and the pressure from the FDA led to the formulation of the first WHO draft text on GMP guidelines in the late 1960s and the GMP guidelines for drugs and medical devices were finalized by the end of 1970s. The European Medicines Agency, known as EMA, was founded in 1995 and it is responsible for assessing and monitoring drug products to maintain and promote public health in the EU. In Europe, the GMP inspections are performed by national regulatory agencies that work under the umbrella of EMA.”
“Unfortunately, the drug scandals do not end here, but several events have led to the revisions of GMP regulations that require stringent monitoring of clinical studies and the use of all human and animal drug products. GMP is constantly evolving to better protect people. That is why GMP is sometimes abbreviated cGMP where the ‘c’ represents current. What was accepted to be good manufacturing practice yesterday may not be accepted today and certainly will not be accepted in the future”, Gustafsson concludes.
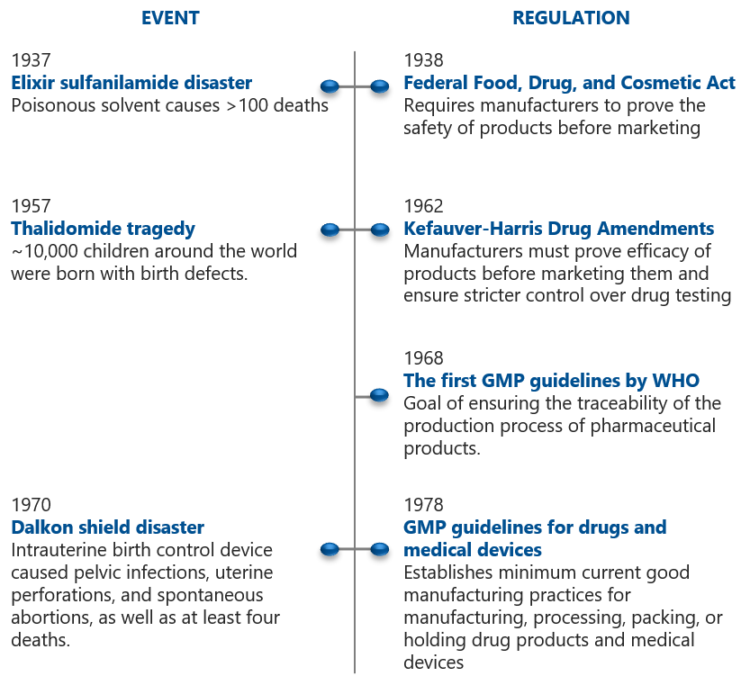
Events and milestones in the development of GMP [1]

Research vs. GMP Production
So, what changes when the project moves from R&D to GMP production? “The differences are all related to quality and traceability,” Eero Mustalahti says. “While the academic setting is characterized by flexibility, there are no shortcuts in implementing GMP. Having a background in research, I can empathize with the drug developers `shock’ when they encounter the strict GMP demands for the first time.”
GMP is not something you decide to do or not do. It is something you must do, because patient safety is top priority in biopharmaceutical manufacturing for human use. Mustalahti explains that although many GMP requirements are general, and thus allow the manufacturer to decide how to best implement them, it also requires that the manufacturer interprets the requirements in a professional way. “Whatever you do, you have to justify based on risk assessment. Mustalahti says. “Failure to comply with GMP can have serious consequences.”
GMP Facility
The production facility needs to be designed for both GMP and biosafety. For example, to meet the cleanroom criteria, the air must be kept free from particulates. “At our facilities the single-pass ventilation design, without any recirculation of air, is an example of this, “Mustalahti says. “While a research lab can be entered through a regular door wearing an ordinary white laboratory coat, entry into a cleanroom takes place through pressured airlocks wearing a cleanroom coverall with a hood and mask. Working in a GMP environment makes you aware of details such as skin flakes, textile fibers and hair that need to be kept away. Of course, the level of requirements depends on the cleanroom classification. In any case, every staff member working in the biopharmaceutical manufacturing must be familiar with the GMP requirements.”
GMP-grade raw materials
In the early development and preclinical stages materials are research-grade, and therefore easily available at lower cost. Once production for clinical study purposes begins, the used raw materials and consumables must be manufactured and documented according to an established quality system. “You cannot just take sodium chloride off the shelf and use it as you could do in academia. The materials and final products must have a documented history. Our job as a CDMO is to source GMP-grade materials. This involves auditing and qualification of material suppliers,” Mustalahti says.
GMP regulations also cover raw material warehousing. Mustalahti explains that raw material receipt, testing, release and storage are strictly controlled. “Storage conditions are controlled, and inventory tracking is in place. Release of material for GMP production can only be done after QA has verified that the raw materials meet all specifications.”
GMP equipment
What makes equipment GMP-compliant? “The short answer is that the equipment needs to be qualified,” Mustalahti says. “Equipment that you can buy, such as bioreactors, analytical instruments, and filling lines, are not GMP-certified in themselves, simply because of the diversity in the biopharmaceutical manufacturing processes. The basic rule is that the equipment used in GMP manufacturing needs to be suitable for its purpose and comply with applicable technical rules. It has to be easy to clean and must not have any negative impact on the quality of the product.”
“Many of the biopharma innovations are coming from academia. Most of our clients have developed processes using equipment that works fine on a small research scale, but not on a large GMP scale. Ultracentrifugation is such a method; it is difficult to scale up. For example, in Viral Vector manufacturing, to process 100 L, you would need a centrifuge the size of a house. Anything more than 10 L is impractical in the GMP setting. Ultracentrifuges are also difficult to clean. The current trend is to replace ultracentrifugation with filtration and chromatography-based purification technologies. Our process development team helps clients with the upscaling to GMP.”
GMP Processes
“When you manufacture GMP biopharmaceuticals, you can manufacture only one product at a time. In academia, you can have a lot of parallel processes, but with GMP it is one room for one product. This of course increases the associated costs,” Mustalahti says.
According to GMP, all operations need to be detailed in writing and documented. When you start GMP production everything has been decided in advance. “Master batch records are an example – they must be written before manufacturing starts, and then you strictly follow that protocol. If e.g. the pH rises during the manufacturing, you cannot just add acid to fix it.” Mustalahti explains. “This is why pre-GMP batches, such as the consistency batch and the engineering batch, are needed. They are run to confirm the process before committing to full-scale GMP manufacturing.”
Analytics
Analytical methods must also follow GMP. Standard Operating Procedures, SOPs, tell the operator exactly how analyzes must be run. “In academia, you can tweak the protocol, like change the flow rate during a chromatographic run, but in GMP you develop a method, and after that, you have to follow the protocol,” Mustalahti says. “The associated software needs to comply with data integrity regulations. For example, analytical results from ddPCR must go into software where the data cannot be manually changed. All data and computer systems used in a GMP facility must be controlled for security and accuracy.”
The GMP-manufactured biopharmaceutical product must meet the QC specifications that are set before production. Examples of specifications are the upper limit for bioburden and endotoxins, the concentration, pH, and osmolality. “If the drug product is outside the QC specifications, quality assurance is not able to issue a certificate of analysis and the qualified person cannot release it. If it were an R&D drug, you could always re-process, re-purify, or concentrate a bit more,” Mustalahti says. “The quality organization of a GMP manufacturer must be independent and not subordinate to another organizational unit. Stability studies are also needed for a Drug Substance and Drug Product, to determine how long it can be used and how it should be stored.”
Summary
GMP came about to ensure that the medicines we take are properly manufactured and tested. These strict GMP regulations have a force of law and they require that manufacturers of biopharmaceuticals for human use take proactive steps to ensure safety and efficacy. GMP covers all aspects of production, from facilities, raw materials, equipment, processes, IT systems to personnel and cleaning. Therefore, adhering to GMP takes more time, costs more, and prohibits spur-of-the-moment changes that might be made in a research laboratory.
[1] https://www.elpro.com/fileadmin/elpro-com/lmn/blog_articles/Historic_Events_and_Milestones_in_the_Development_of_GMP.pdf


